Jump to:
Molecular mechanisms underlying the regulation of CRAC channel and SOCE by ORAI and STIM proteins
Primary immunodeficiency (PID) due to mutations in ORAI1 and STIM1 genes
CRAC channels in innate and adaptive immunity to infection
CRAC channels in immunological tolerance and autoimmune diseases such as multiple sclerosis (MS) and inflammatory bowel disease
CRAC channels in cancer and antitumor immunity
Ca2+ signals and immunometabolism
Characterization of novel ion channels and transporters in immune function
Molecular mechanisms of CRAC channel regulation by ORAI and STIM proteins.
CRAC channels are formed by a family of three well conserved integral membrane proteins called ORAI1, ORAI2 and ORAI3 (so named after the Horae from Greek mythology – see fun facts). CRAC channel currents were first recorded in the late 1980s, but their molecular identity remained elusive for almost 20 years. In 2006, using positional cloning by linkage analysis in CRAC channel-deficient patients and siRNA screens in Drosophila S2 cells, we and others identified ORAI1 and its two homologues as essential components of the CRAC channel (1). We demonstrated that ORAI1 is the pore-forming subunit of the CRAC channel by mutagenesis and electrophysiological analysis of the biophysical properties of ORAI proteins (2). These ground breaking studies identified the long elusive nature of the CRAC channel and enabled many labs to study its molecular regulation and investigate its physiological roles in various tissues in health and disease. In subsequent studies we showed that ORAI1 channel function is modulated by glycosylation and phosphorylation and identified a critical coiled-coil domain in STIM1 that is necessary and sufficient for CRAC channel activation (3). We recently showed that ORAI2, which functions as a Ca2+ channel when overexpressed, forms heteromeric CRAC channel complexes with ORAI1, in which ORAI2 modulates the magnitude of SOCE to control T cell-mediated immune responses (4). The Feske lab uses a variety of molecular biology tools including gene-editing and knockout mice as well as electrophysiological and fluorescent imaging techniques (including TIRF and FRET microscopy) to investigate the molecular mechanisms and physiological roles of ORAI1, ORAI2, ORAI3, STIM1 and STIM2 proteins.
- Feske S et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006; 441:179-85.
- Prakriya M et al. Orai1 is an essential component of the CRAC channel pore. Nature 2006; 443:230-3.
- Kawasaki T et al. A minimal regulatory domain in the C terminus of STIM1 binds to and activates ORAI1 CRAC channels. Biochem Biophys Res Commun. 2009, 385:49–54.
- Vaeth M et al. ORAI2 modulates store-operated calcium entry (SOCE) and T cell-mediated immune responses. Nature Comm 2017; 8:14714.
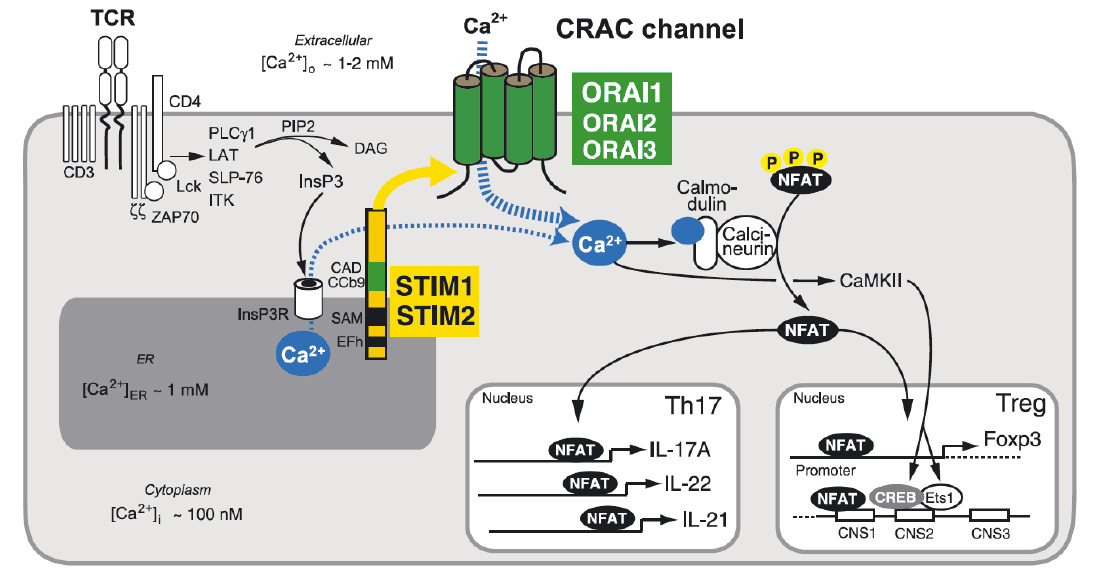
Store-operated Ca2+ entry (SOCE) and Ca2+ signaling in T cells. T cell receptor (TCR) stimulation activates protein tyrosine kinases such as lck and ZAP-70, which results in activation of phospholipase C (PLC) γ1. PLCγ1 catalyzes the hydrolysis of PIP2 into inositol-1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). (Note that binding of G protein coupled chemokine receptors can also cause production of IP3 via activation of PLCb). IP3 binds to and opens IP3 receptors (IP3R), which release Ca2+ from ER Ca2+ stores. A decrease in the ER Ca2+ concentration is sensed by STIM1 and STIM2 resulting in the activation of CRAC channels in the plasma membrane that are formed by ORAI proteins. The resulting Ca2+ influx, called SOCE, activates Ca2+-dependent enzymes such as calcineurin and transcription factors such as NFAT, NF-kB and CREB in various T cell subsets. Ca2+ concentrations in- and outside the cell are indicated.
From: Shaw PJ & Feske S. 2012 J. Physiology 590:4157-67.
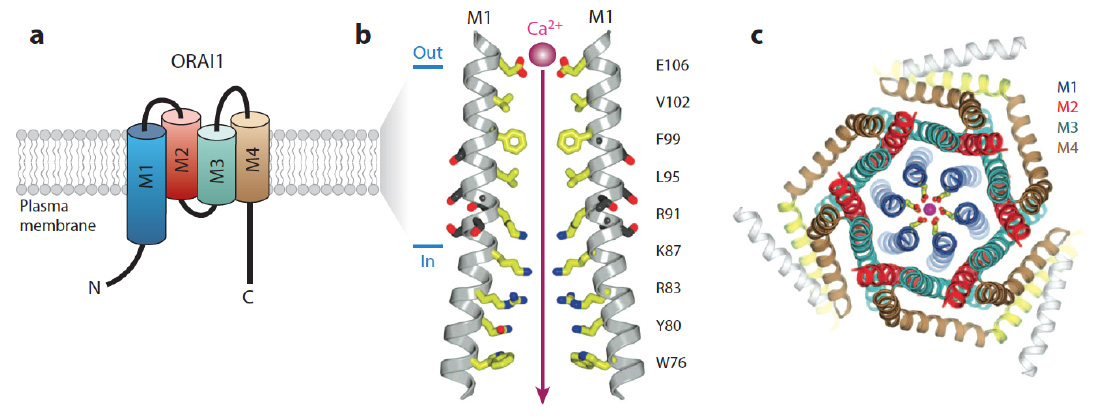
ORAI structure. The CRAC channel is formed by ORAI proteins, which are integral membrane proteins. (a) Membrane topology of ORAI1. Each ORAI1 protein has four transmembrane domains (M1-4), intracellular N- and C-termini and two extracellular loops. (b) The pore of the Drosophila Orai (dOrai) channel. M1 lines the conduction pathway for Ca2+. Two M1 alpha-helices from two separate dOrai subunits are shown with amino acid side chains protruding into the pore indicated in yellow. Amino acid residue numbers refer to human ORAI1. Glutamate (E) 106 at the outer end of the pore is the Ca2+ binding site in the selectivity filter of the CRAC channel that determines its preference for conducting Ca2+ over other divalent or monovalent cations. R91 is mutated in patients with abolished CRAC channel function. (c) Hexameric assembly of dOrai subunits shown in an orthogonal view from the extracellular side. The colors for each transmembrane helix in this panel are the same as in panel a; E106 is depicted in yellow and a Ca2+ ion in magenta. (Panels b and c adapted from Hou et al. Science. 2012; 338:1308-13).
From: Feske et al. 2015 Ann Rev Imm 33:291-353.
.
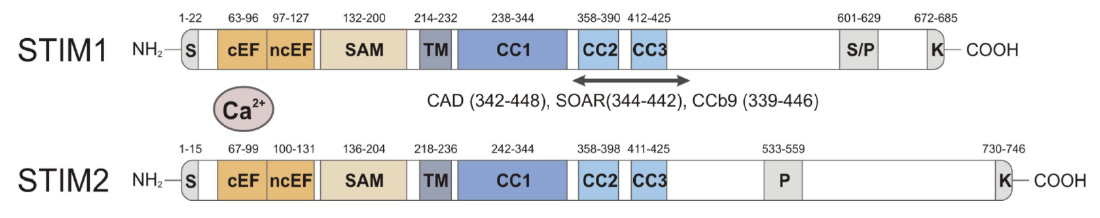
Domain structure of STIM1 and STIM2. CRAC activation (CAD) domain (alternative names are STIM1-ORAI activating region (SOAR) and coiled-coil domain containing fragment b9 (CCb9), which contains 2 coiled-coil domains, CC2 and CC3. The CAD/SOAR/CCb9 domain of STIM1 is necessary and sufficient to activate ORAI1 and SOCE.
From: Shaw PJ & Feske S. Cell Mol Life Sci. 2013; 70:2637-56
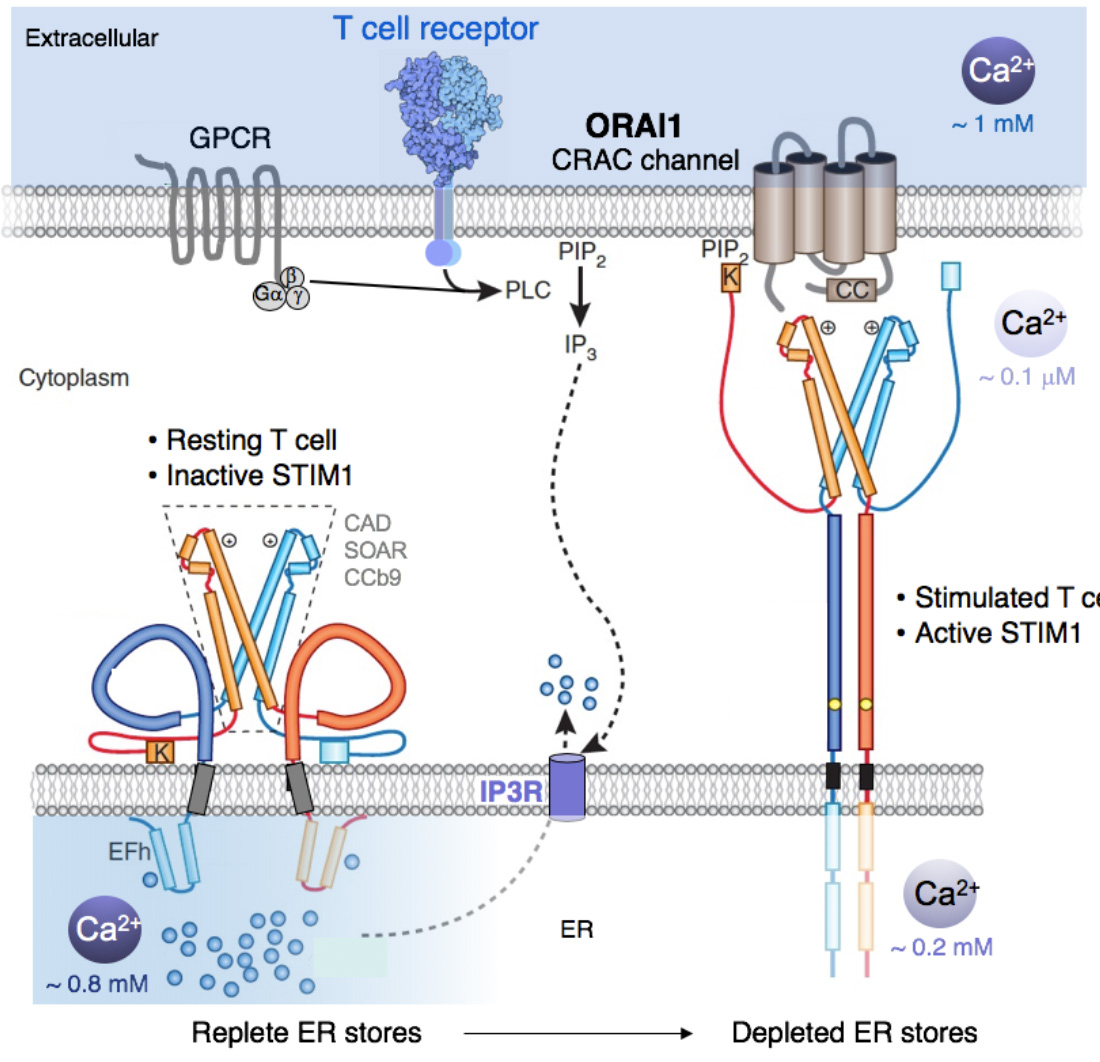{kind=link}
Molecular choreography of STIM1 activation and binding to ORAI1 channels. T cell stimulation results in the production of IP3 and IP3R-mediated release of Ca2+ from ER Ca2+ stores. The resulting fall in [Ca2+]ER from ~ 0.8 mM to below 0.4 mM triggers the dissociation Ca2+ ions from an EF-hand in the N terminus (NT) of STIM1, which results in the unfolding of the NT of STIM1 and the dimerization of the NT of neighbouring STIM1 molecules. This triggers conformational changes in the C terminus (CT) of STIM1 that result in its elongation and allow it to straddle the gap between the ER membrane and the plasma membrane (PM) within ER-PM junctions, which are regions of the cell in which the ER comes in close proximity to the PM. In these junctions, STIM1 binds to negatively charged phospholipids in the PM via a lysine-rich (K) domain and to ORAI1 via its CRAC activation (CAD) domain to induce CRAC channel activation. Note the ~ 10,000 fold concentration gradient between cytosolic Ca2+ and Ca2+ in the ER and extracellular space.
Primary immunodeficiency due to mutations in ORAI1 and STIM1 genes.
My lab identified the first patients with autosomal recessive loss-of-function mutations in ORAI1 and STIM1 genes (1,2). These patients suffer from a Primary Immunodeficiency (PID) syndrome we defined and named CRAC channelopathy (3). It is defined by a spectrum of immunological and non-immunological symptoms including (a) combined immunodeficiency (CID) with recurrent and chronic infections, (b) autoimmunity due to autoantibody-mediated hemolytic anemia and thrombocytopenia, (c) muscular hypotonia with atrophy of type II muscle fibers and (d) anhidrotic ectodermal dysplasia. Patients with CRAC channelopathy present with CID despite largely normal lympho- and myelopoiesis as well as serum immunoglobulin levels. However, several important effector functions of T and NK cells are impaired in the absence of functional CRAC channels including T cell proliferation, cytokine production and cytotoxic effector functions. My lab works with pediatricians around the world to study patients with CRAC channelopathy to understand the causes of their CID and non-immunological symptoms. Naturally occuring LOF mutations in ORAI1 and STIM1 genes that interfere with protein function provide new insights into the molecular regulation of CRAC channel function (4). To analyze in more detail the role of CRAC channels in immunity, anhidrosis (the inability to sweat) (5) and amelogenesis imperfecta (defective dental enamel development) (6,7) we are using Orai and Stim knockout mice and collaborate with a variety of labs at NYU and elsewhere.
- Feske S et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006; 441:179-85.
- Picard C et al. STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. New England J Medicine. 2009 360:1971-80.
- Feske S. CRAC channelopathies. Pflügers Archive European Journal of Physiology 2010, 460:417-35.
- Maus M et al. Missense mutation in immunodeficient patients shows the multifunctional roles of coiled-coil domain 3 (CC3) in STIM1 activation. Proc Natl Acad Sci U S A. 2015 112:6206-11.
- Concepcion A et al. Store-operated Ca2+ entry regulates Ca2+-activated chloride channels and eccrine sweat gland function. J Clin Invest 2016, 126:4303-4318.
- Eckstein M et al. Store-operated Ca2+ entry (SOCE) controls ameloblast cell function and enamel development. Journal of Clinical Investigation Insight 2017, Mar 23;2(6):e91166
- Eckstein M et al. Differential regulation of Ca2+ influx by ORAI channels mediate enamel mineralization. Science Signaling 2019 Apr 23;12(578).
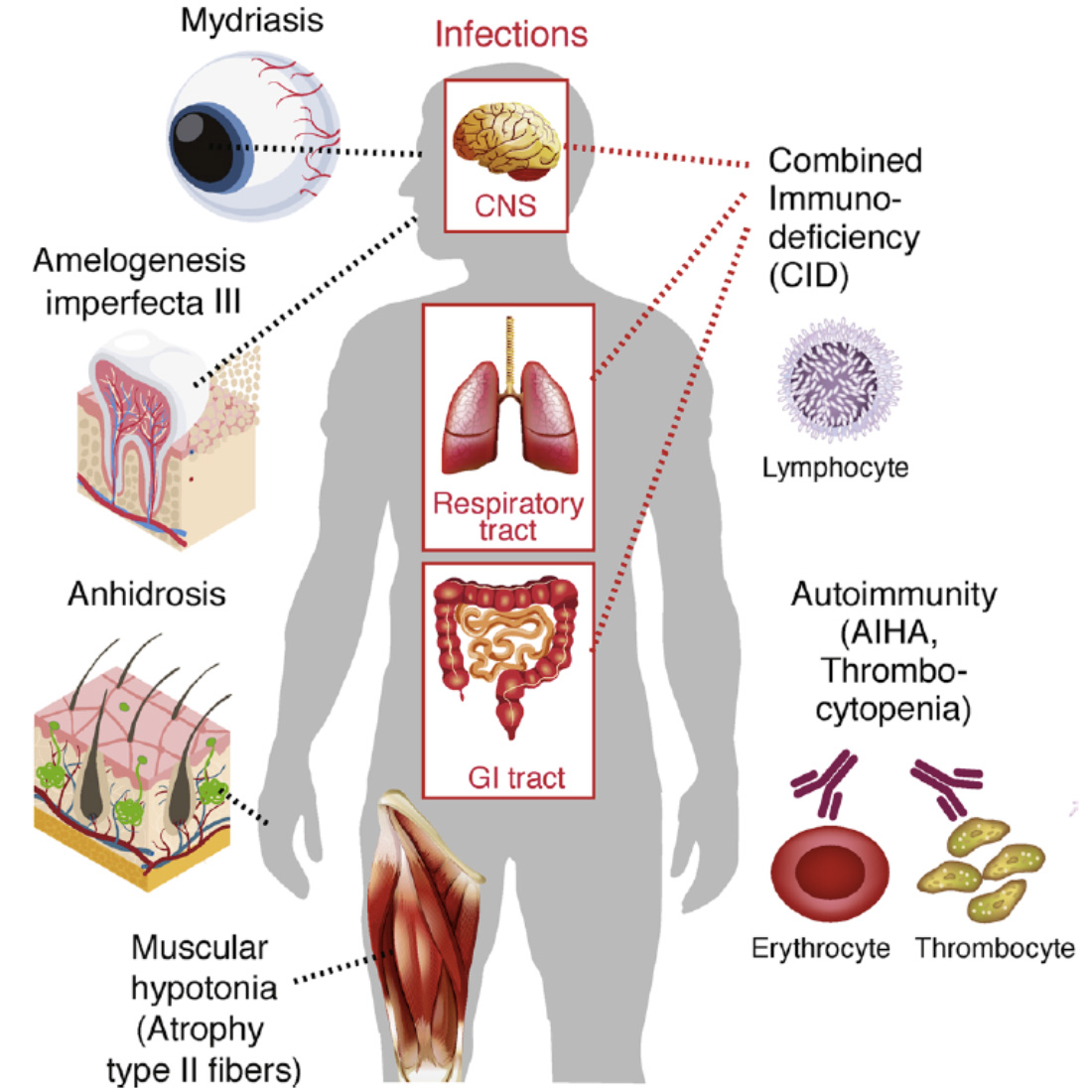
CRAC channelopathy in human patients. Loss-of-function (LOF) mutations in ORAI1 (OMIM 612782) and STIM1 (OMIM 612783) genes result in (1) combined immunodeficiency with chronic, often lethal infections with bacterial, viruses and fungi, (2) autoimmunity, (3) anhidrotic ectodermal dysplasia (EDA) and (4) muscular hypotonia. AIHA, autoimmune hemolytic anemia.
From: Feske S. Cell Calcium. 2019 Jun;80:112-116.
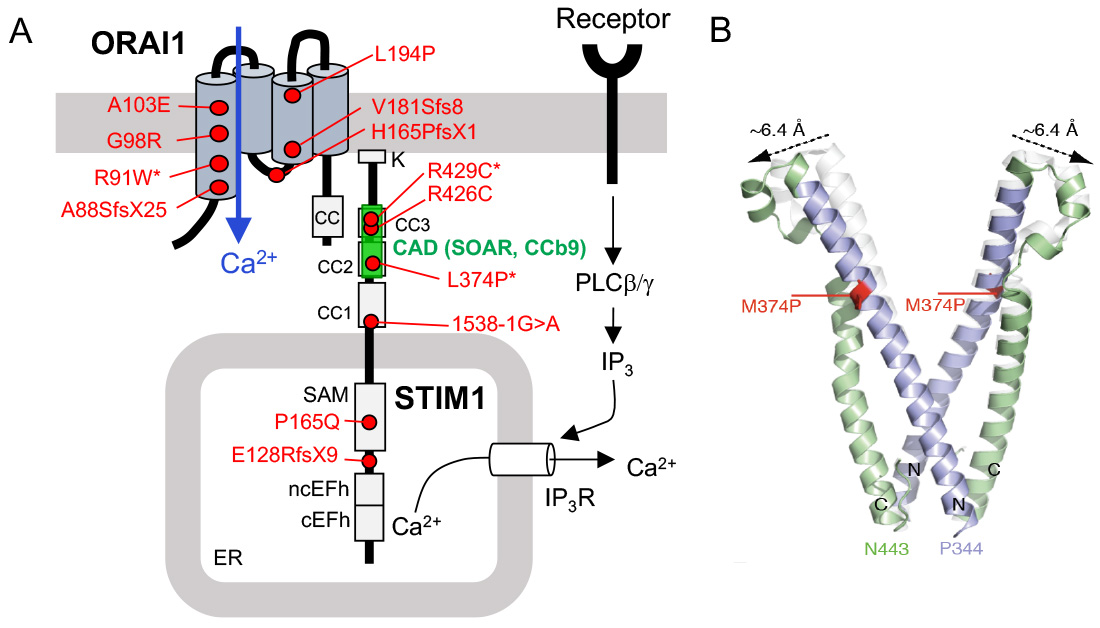
CRAC channelopathy in human patients due to loss-of-function (LOF) mutations in ORAI1 and STIM1 genes. (A) Known mutations that abolish protein function but not expression are indicated by an *. The CRAC activation domain (CAD, which is also named SOAR or CCb9) is essential for STIM1 binding to ORAI1. (B) Structure model of wildtype (white) and mutant (p.L374P) CAD domain of STIM1 (green and purple). The mutation shifts the apex position of the CAD domain, potentially interfering with the binding of STIM1 to ORAI1.
From: Lacruz R & Feske S. Ann N Y Acad Sci. 2015, 1356:45-79 and Kahlfuss S. et al. (submitted).
CRAC channels in innate and adaptive immunity to infection
The importance of CRAC channels for immunity to infection is evident from the Primary Immunodeficiency (PID) of patients with inherited loss-of-function mutations in ORAI1 or STIM1 genes. Because these patients are very rare, we are using gene-targeted mice with deletion of Stim1, Stim2, Orai1, Orai2 and Orai3 genes to investigate the mechanisms by which CRAC channels regulate innate and adaptive immunity to infection with a variety of pathogens. We found that abolished Ca2+ influx in STIM1 and STIM2-deficient mice severely impairs T cell-mediated immune responses to viral infection with LCMV and the maintenance of CD8+ T cell memory (1). In CD4+ T cells, SOCE is also critical for controling immunity to chronic infection with M. tuberculosis by regulating cell death, cytokine production and the inflammation accompanying unresolved, chronic infections (2). Although immunolglobulin levels are normal in CRAC channel deficient patients and mice, they fail to produce a variety of pathogen specific antibodies after vaccination or infection. We found that SOCE is critical for the differentiation and function of CD4+ follicular helper T cells (TFH cells), which help B cells to mature and to produce high-affinity antigen-specific antibodies (3). In the absence of STIM1 and STIM2, the humoral immune response to viral infections with LCMV and influenza is impaired. These ongoing studies have elucidated essential roles of CRAC channels in adaptive immunity and the mechanisms underlying immunodeficiency and susceptibility to infections in ORAI1 and STIM1-deficient patients.
- Shaw PJ et al. CD4⁺ and CD8⁺ T cell-dependent antiviral immunity requires STIM1 and STIM2. J Clin Invest 2014, 124:4549-63.
- Desvignes L et al. STIM1 controls T cell-mediated immune regulation and inflammation in chronic infection. J Clin Invest 2015, 125:2347-62.
- Vaeth M et al. Store-operated Ca2+ entry in follicular T cells controls humoral immune responses and autoimmunity. Immunity 2016, 44:1350-64.
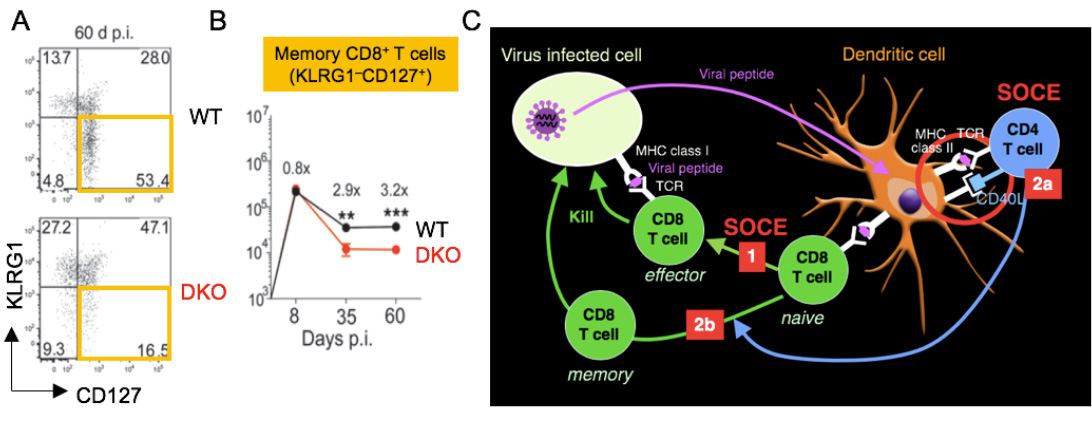
SOCE regulates CD8+ T cell immunity to viral infection. (A,B) Reduced frequencies of KLRG1- CD127+ memory CD8+ T cells at 60 days after infections with LCMV. (C) SOCE is required for the expansion of effector CD8+ T cells (1), the priming of CD4 T cells by DC via CD40L (2a) as well as the maintenance and function of virus-specific memory CD8+ T cells (2b).
From Shaw et al. 2014, J Clin Invest. 124:4549-63.
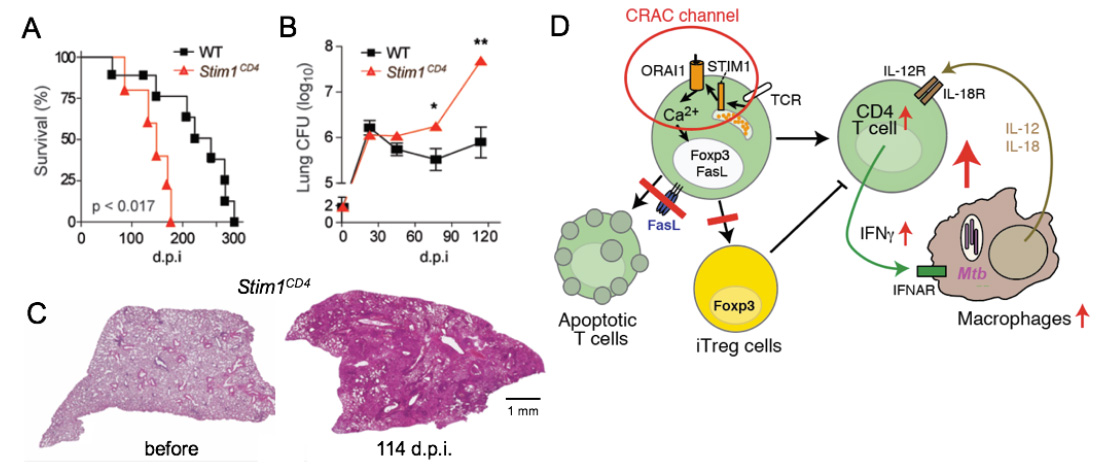
SOCE controls immunity to chronic infection with Mycobacterium tuberculosis (Mtb). (A-C) Mice with conditional deletion of STIM1 in T cells succumb to Mtb infection significantly faster than WT mice (A), have increased Mtb burdens in their lungs (B) and massive pulmonary inflammation (C). Model how SOCE normally controls chronic infection and keeps inflammation in check. In the absence of SOCE, T cell apoptosis and the differentiation of CD4 T cells into iTreg cells is impaired, both resulting in increased numbers and activity of T cells. During the course of infection, SOCE-deficient T cells become more sensitive to IL-12 and IL-18 stimulation, resutling in the Ca2+ independent production of excessive amounts of IFN@gamma;, which drives myeloid cell expansion and inflammation.
From: Desvignes L, J Clin Invest 2015, 125:2347-62.
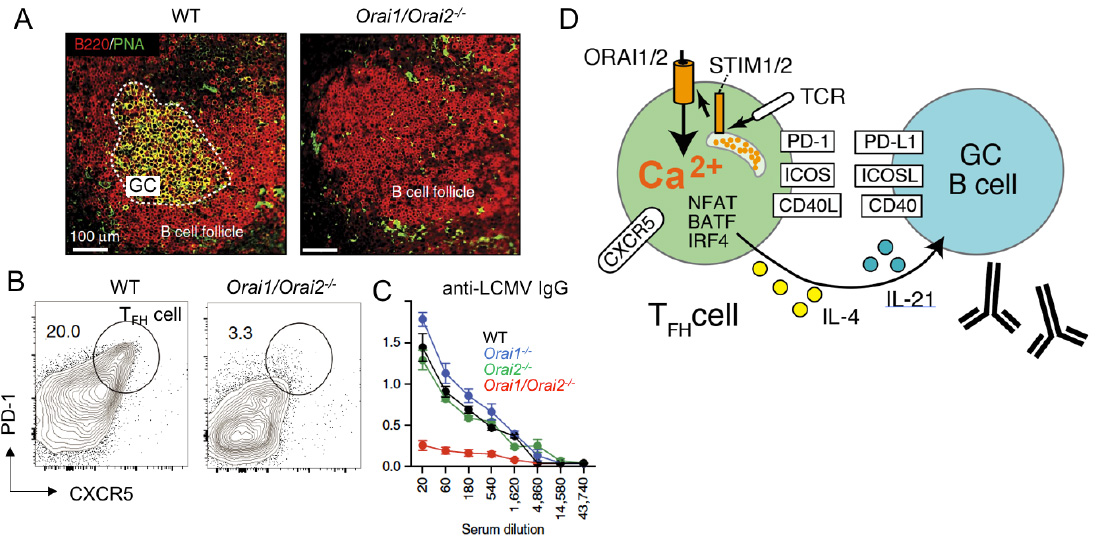
SOCE in follicular T helper (TFH) cells controls humoral immunity. (A-C) Lack of SOCE in T cells of Orai1-/- Orai2-/- mice results in failed germinal center (GC) formation (A), TFH cell differentiation (B) and IgG antibody production (C) after infection with lymphocytic choriomeningitis virus (LCMV). (D) SOCE regulates expression of many transcription factors, cytokines and surface receptors on TFH cells that are critical for the maturation of GC B cells and antibody production.
From: Vaeth M et al. 2017, Nature Comm 8:14714
CRAC channels in immunological tolerance and autoimmunity.
Ca2+ influx and CRAC channels are not only essential for immunity to infection, they also control immunological tolerance and thus autoimmunity and inflammation. Patients with LOF mutations in ORAI1 and STIM1 develop autoantibodies against nuclear antigens (ANA), red blood cells and platelets that cause autoimmune hemolytic anemia (AIHA) and thrombocytopenia. The simultaneous presence of immunodeficiency and autoimmunity is common to many forms of PID, but the mechanisms how SOCE controls immunological tolerance are specific to CRAC channelopathy. We demonstrated that SOCE is required for the development and function of immunosuppressive Foxp3+ Treg cells in the thymus by using mice with conditional deletion of Stim1 and Stim2 genes (1). The numbers of Treg cells are reduced in the blood of most ORAI1 or STIM1-deficient patients and in lymphoid organs of Stim1 and Stim2-deficient mice. SOCE is of particular importance for the differentiation of follicular Treg (TFR) cells, which suppress the spontaneous maturation of autoreactive B cells in germinal centers of lymph nodes and spleen, thus preventing the production of autoantibodies (2). Conditional deletion of CRAC channel genes in T cells or Treg cells results in the production of a spectrum of autoantibodies including anti-dsDNA and antinuclear antibodies (ANA). SOCE is furthermore required for the differentiation of thymus-derived Treg cells into tissue-resident Treg cells that control immune homeostasis in organs (3) and naive CD4 T cells into peripheral Treg (pTreg) cells, for instance in the context of chronic infections (4).
Besides Treg cells, Ca2+ influx through CRAC channels controls the effector functions of T helper 1 (Th1) and Th17 cells, which mediate inflammation in many forms of autoimmune disease. Suppression of SOCE in Th1 and Th17 cells abolishes the production of IFNγ, IL-17, GM-CSF and other proinflammatory cytokines. Consequently, deletion of Stim1, Stim2 or Orai1 in T cells or pharmacological inhibition of CRAC channels protects mice from experimental autoimmune encephalomyelitis (EAE), an animal model of Multiple Sclerosis (MS), inflammatory bowel disease (IBD) and Th17-mediated skin and lung inflammation (5-8). Although immunosuppressive Treg cells and proinflammatory Th1/Th17 cells both require SOCE, we find that Th1/Th17-mediated autoimmunity is much more susceptible to partial suppression of SOCE, whereas Treg-dependent immune regulation is compromised only when SOCE is almost completely abolished. These different quantitative SOCE requirements of pro- and anti-inflammatory CD4+ T cells suggest that a therapeutic window for CRAC channel inhibition exists, which may allow for the selective inhibition of Th1/Th17, but not Treg cells, to suppress autoimmune inflammation.
- Oh-hora M et al. Dual role of ER Ca2+ sensors Stim1 and Stim2 in T cell activation and tolerance. Nature Immunol 2008; 9:432-43. * co-corresponding authors
- Vaeth M et al. Store-operated Ca2+ entry in follicular T cells controls humoral immune responses and autoimmunity. Immunity 2016, 44:1350-64.
- Vaeth M et al. Tissue resident and follicular Treg cell differentiation is regulated by CRAC channels. Nature Commun. 2019 Mar 12;10(1):1183.
- Desvignes L et al. STIM1 controls T cell-mediated immune regulation and inflammation in chronic infection. J Clin Invest 2015, 125:2347-62.
- Ma J et al. T cell specific deletion of STIM1 and STIM2 protects mice from EAE by impairing effector functions of TH1 and TH17 cells. Eur J Immunol. 2010, 40:3028-42.
- McCarl CA et al. Store-operated Ca2+ entry through ORAI1 is critical for T cell mediated autoimmunity and allograft rejection. J Immunol. 2010, 185:5845-58.
- Kaufmann U et al. Selective ORAI1 Inhibition Ameliorates Autoimmune Central Nervous System Inflammation by Suppressing Effector but Not Regulatory T Cell Function. J Immunol. 2016, 196:573-85.
- Kaufmann U et al. Calcium Signaling Controls Pathogenic Th17 Cell-Mediated Inflammation by Regulating Mitochondrial Function. Cell Metabolism 2019 Feb 14. pii: S1550-4131(19)30019-1.
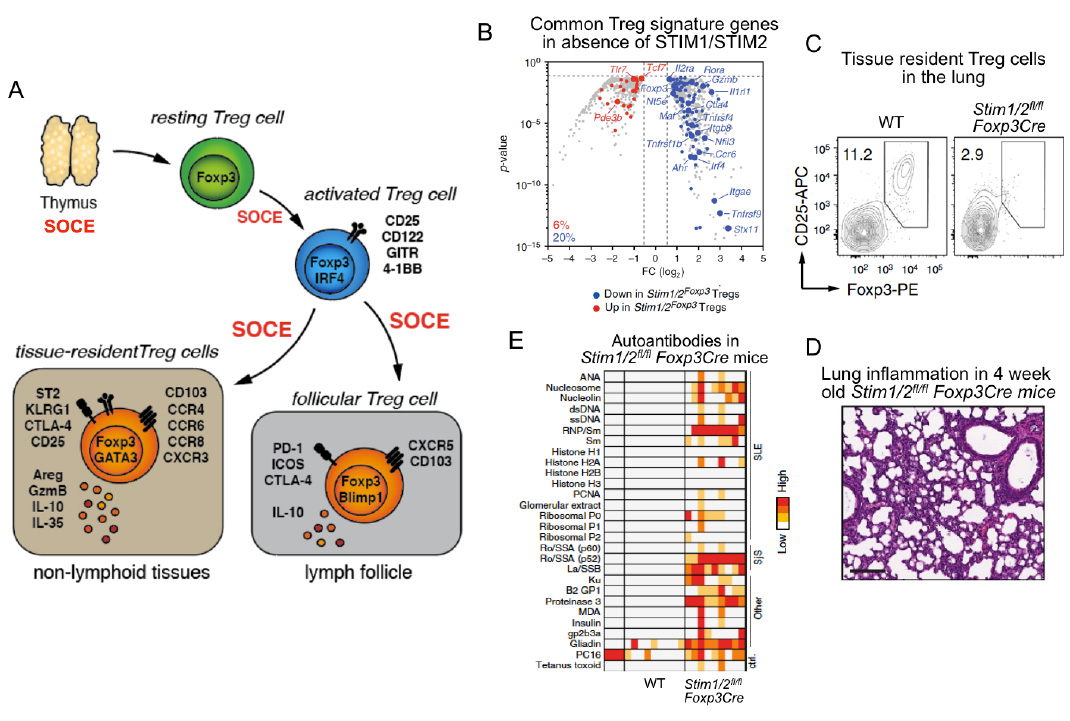
SOCE through CRAC channels controls regulatory T cells (Treg). SOCE is required for the development of Foxp3+ TReg cells in the thymus (see Oh-hora 2008 Nat. Imm). Once in the periphery, SOCE is required for Treg cell activation and promoting Treg-specific gene expression(panel B). SOCE is also required for the differention of Treg cells into tissue-resident Treg cells and the prevention of inflammtion in organs such as lung (panels C,D), liver, kidney and skin. Furthermore, SOCE promotes the differentiation of Treg cells into follicular Treg cells that control the germinal center (GC) reaction in lymph nodes and spleen. In the absence of SOCE in Treg cells, Stim1fl/fl Stim2fl/fl Foxp3Cre mice develop a plethora of autoantibodies and multiorgan inflammation (panel E).
From: Vaeth M et al. Nature Communications 2019, 10(1):1183.
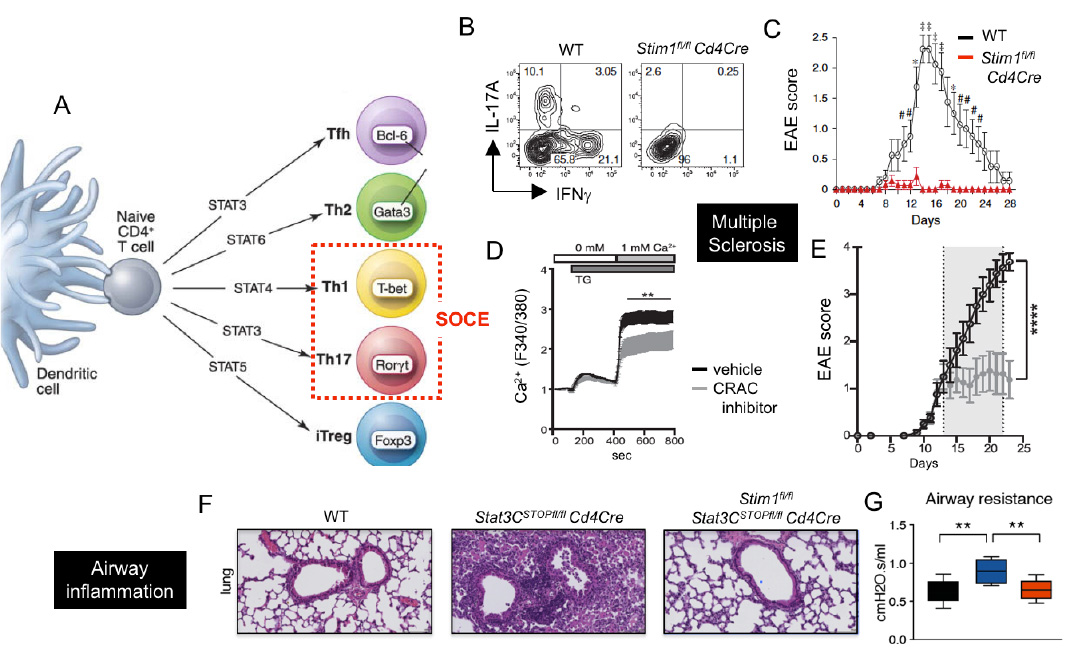
CRAC channels control Th1 and Th17 cell function and autoimmune inflammation. (A) SOCE is required for the differentiation and function of Th1 and Th17 cells. (B-E) CRAC channels in T cells promote CNS inflammation in the EAE model of multiple sclerosis (MS). (B,C) T cell specific deletion of STIM1 abolishes IL-17A and IFN@gamma; production and prevents EAE. (D,E) Treatment of mice with the CRAC channel inhibitor AMG1 inhibits SOCE (D) and stops EAE progression (E). (F-G) Deletion of STIM1 prevents pulmonary inflammation and dysfunction in a STAT3-driven model of pathogenic Th17 cell development and autoimmune inflammation.
From: Ma et al. 2010, Eur J Immunol. 40:3028-42 (B,C), Kaufmann et al. 2016, J Immunol. 196:573-85 (D,E), Kaufmann et al. 2019, Cell Metab. 29:1104-1118 (F,G)
CRAC channels in cancer and antitumor immunity
CRAC channel function and SOCE are critical for the ability of cytotoxic lymphocytes including CD8+ T cells and NK cells to kill virus-infected and tumor cells in vitro. CD8+ T cells lacking functional CRAC channels have reduced levels of death receptors, lytic granule exocytosis and expression of IFN@gamma; and TNFα. As a consequence, CD8+ T cells lacking SOCE due to genetic deletion of Stim1 and Stim2 genes fail to control tumor growth in vivo (1). Besides antitumor immunity, CRAC channels appear to directly regulate cancer growth and metastasis as shown by numerous studies of their role in melanoma, breast and prostate cancer and other malignancies. In hematopoietic malignancies, their role is less well investigated. We demonstrated that transcript levels of the principal CRAC channel components ORAI1 and STIM1 were highest in human T cell acute lymphoblastic leukemia (T-ALL) compared to 40 other cancers, suggesting that SOCE is important in T-ALL (2). T-ALL is commonly associated with activating mutations in the Notch1 signaling pathway and using a Notch1-dependent mouse model of T-ALL, we found that deletion of STIM1 and STIM2 in leukemic cells abolishes SOCE and significantly prolongs the survival of mice. The survival advantage was unrelated to leukemic cell burdens but associated with a markedly reduced necroinflammatory response in leukemia-infiltrated organs in the absence of STIM1 and STIM2. Thus, SOCE in leukemic T lymphoblasts promotes the inflammation of leukemia-infiltrated organs and thereby negatively impacts survival in T-ALL. Our study sheds new light on how leukemic T cells influence their tumor microenvironment, which may have important implications for leukemia treatment.
- Weidinger C et al. STIM1 and STIM2 mediated Ca2+ influx regulates antitumor immunity by CD8+ T cells. EMBO Mol Med. 2013, 5:1311-21.
- Saint Fleur-Lominy S et al. Store-operated Ca2+ entry mediates cancer-induced inflammation in T cell acute lymphoblastic leukemia. Cell Reports. 2018, 24:3045-3060.
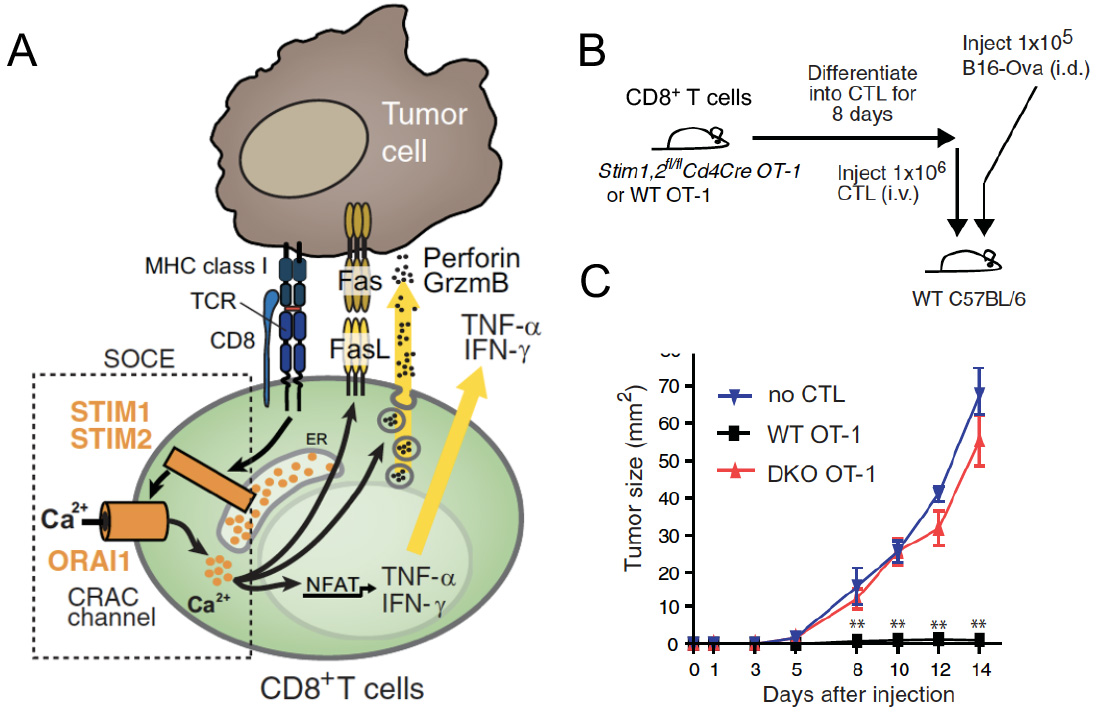
SOCE in cytotoxic CD8+ T lymphocyte (CTL) function and antitumor immunity. (A) TCR stimulation results in the activation of STIM1 and STIM2, opening of CRAC channels formed by ORAI proteins and SOCE. The subsequent intracellular Ca2+ increase mediates IFN@gamma; and IFN@alpha; production, FasL expression and degranulation of CTLs, which are required for antitumour immunity by CD8+ T cells. (B,C) Deletion of STIM1 and STIM2 in CTL prevents their ability to prevent the engraftment of tumor cells (B16 melanoma) after adoptive T cell transfer resulting in rapid tumor growth.
From: Weidinger C et al. EMBO Mol Med. 2013, 5:1311-21
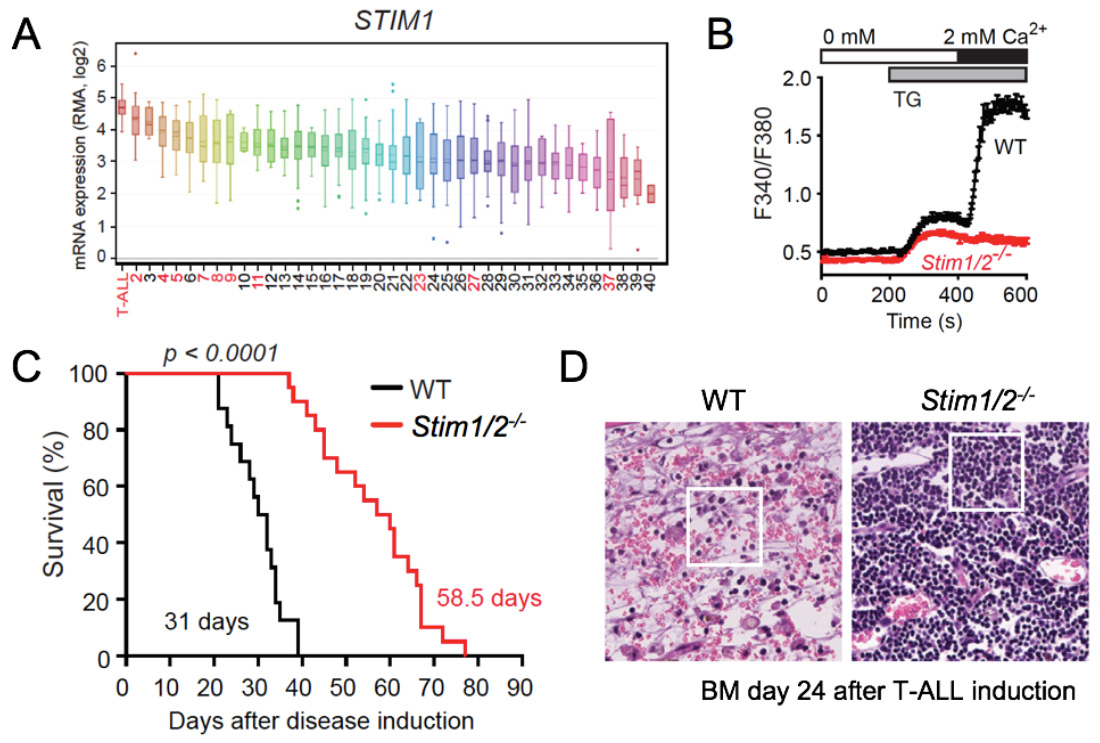
STIM1 and STIM2 promote leukemia progression by mediating cancer-induced inflammation in T cell acute lymphoblastic leukemia (T-ALL). (A) STIM1 and ORAI1 (not shown) are most highly expressed in T-ALL lymphoblasts compared other human tumors and hematological malignancies (in red). (B) Abolished SOCE in leukemic T lymphoblasts from Stim1/2-/- mice. (C) Prolonged survival of Stim1/2-/- mice in a Notch1-driven model of T-ALL. (D) H&E staining of BM (femur) from WT and Stim1/2-/- mice 24 days after T-ALL induction shows extensive necroinflammatory response in WT, whereas no signs of necrosis are apparent in Stim1/2-/- mice whose organs are filled with monotonous lymphoid cells without inflammation.
From: Saint Fleur-Lominy S et al. Cell Reports. 2018, 24:3045-3060.
Ca2+ influx through CRAC channels controls several metabolic pathways in T cells
The metabolic regulation of immune responses has attracted a lot of attention in recent years. We found that CRAC channels and SOCE are key regulators of several metabolic pathways in immune and non-immune cells. One of the most profound roles of SOCE is to control to expression of nuclear encoded mitochondrial genes, including many components of the electron transport chain (ETC). In the absence of SOCE in T cells from patients with ORAI1 or STIM1 mutations and mice with deletion of Stim1 and Stim2 genes mitochondrial respiration and oxidative phosphorylation (OXPHOS) are severely impaired (1). We showed that SOCE controls the mitochondrial function of pathogenic Th17 cells, which play important roles in many autoimmune and inflammatory diseases. STIM1-deficient pathogenic Th17 cells have significantly reduced expression of many ETC components resulting in impaired OXPHOS. STIM1 deletion or inhibition of OXPHOS is associated with impaired pathogenic Th17 cell function in vivo resulting in attenuated skin and lung inflammation in a STAT3-driven model of Th17 disease (2). Besides mitochondrial function, SOCE controls the glycolytic metabolism of activated T cells. It does so by controlling the transcriptional regulation of glycolytic enzymes, glucose transporters and transcription factors like Myc, IRF4 and HIF1 that act as master regulators of glycolytic metabolism (1). In addition, SOCE regulates the PI3K-AKT kinase-mTOR nutrient sensing pathway at a posttranslational level. Many of these SOCE dependent changes are mediated by the activation of the transcription factor NFAT as deletion of NFAT1 and NFAT2 in T cells results in severely impaired glycolysis. As a consequence of abolished SOCE, T cells show a profound defect in proliferation and cell cycle progression in vitro and in vivo. We conclude that SOCE controls a critical metabolic checkpoint at which T cells assess adequate nutrient supply to support their clonal expansion. In non-immune cells, we found that SOCE also plays an important role in controlling lipolysis and the transcriptional reprogramming of cells to lipid metabolism (3). Lack of SOCE results in the accumulation of pathological amounts of lipid droplets in cells in vitro and in vivo as SOCE is critical for the mobilization of fatty acids from lipid droplets, the expression and activation of neutral lipases that mediate lipolysis, and mitochondrial fatty acid oxidation (FAO). These changes are due in part to the SOCE-dependent expression of transcriptional regulators of lipid metabolism, PGC-1α and PPARα. Ongoing and future studies will address how SOCE regulates metabolic pathways in immune cells and thereby adaptive and innate immunity.
- Vaeth M et al. Store-operated Ca2+ entry controls clonal expansion of T cells through metabolic reprogramming. Immunity 2017, 47:664-679
- Kaufmann U et al. Calcium Signaling Controls Pathogenic Th17 Cell-Mediated Inflammation by Regulating Mitochondrial Function. Cell Metabolism 2019, 29:1104-1118.
- Maus M et al. Store-Operated Ca2+ Entry Controls Induction of Lipolysis and the Transcriptional Reprogramming to Lipid Metabolism. Cell Metabolism 2017. 25:698-712.
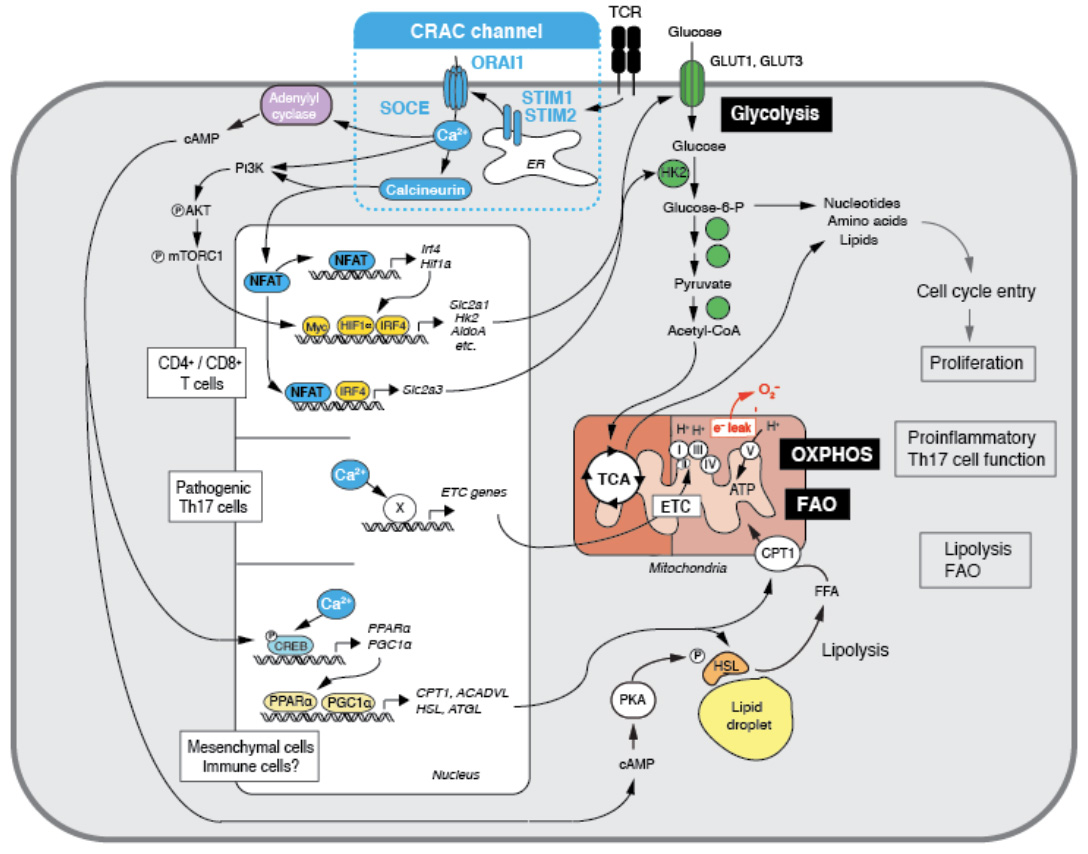
Ca2+ influx through CRAC channels controls several metabolic pathways in T cells. (1) SOCE controls the upregulation of glycolytic enzymes and glucose transporters via regulating the activity and expression of the transcription factors NFAT, myc, IRF4 and HIF-1a. Glycolysis is required for the proliferation and clonal expansion of T cells (from: Vaeth M et al. Immunity 2017, 47:664-679). (2) SOCE controls mitochondrial function in Th17 cells and other CD4+ cells as well as mesecnchymal cells through the expression of electron transport chain (ETC) genes, thus enabling oxidative phosphorylation (OXPHOS) and the pathogenic function of Th17 cells and multiorgan inflammation (from: Kaufmann U et al. Cell Metabolism 2019, 29:1104-1118). (3) SOCE controls the transcriptional reprogramming mesenchymal cells to support lipid metabolism, resulting in enhanced lipolysis and mitochondrial fatty acid oxidation (FAO) (from: Maus M et al. Cell Metabolism 2017. 25:698-712).
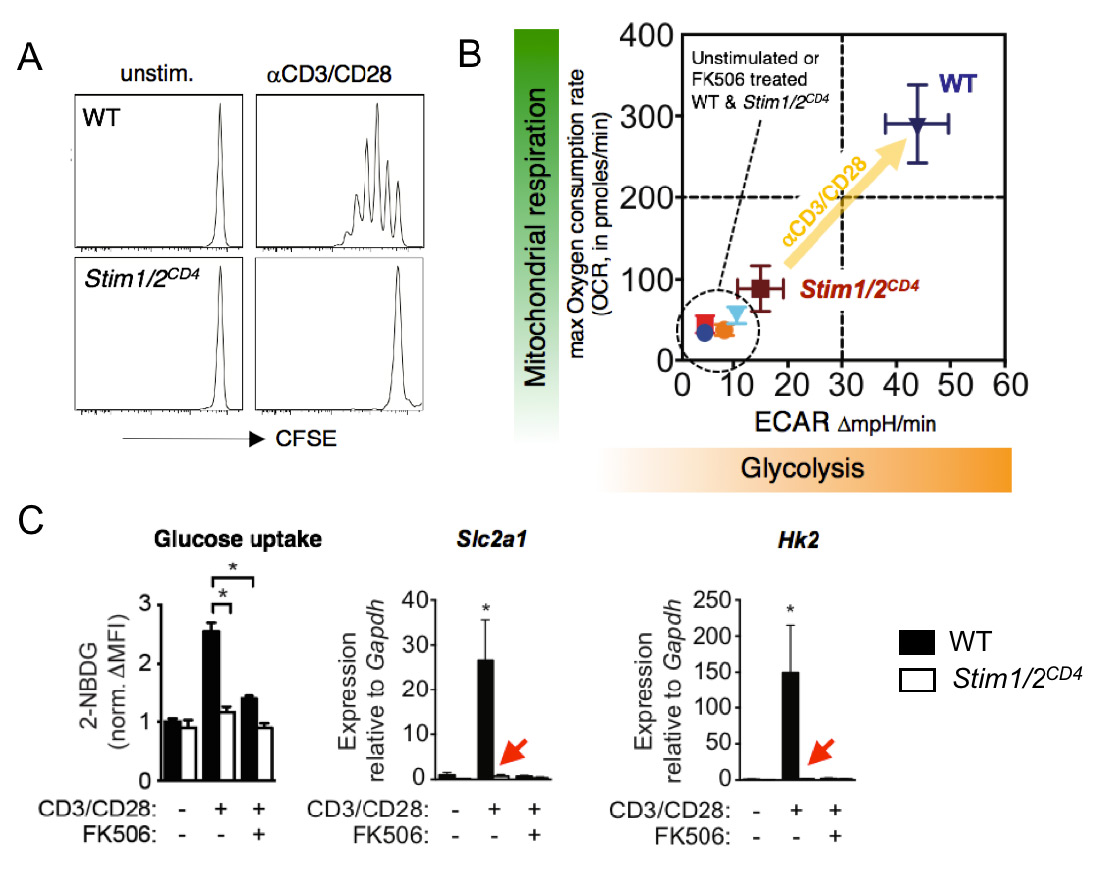
The CRAC channel activating proteins STIM1 and STIM2 are essential for aerobic glycolysis and proliferation of T cells. (A) Impaired TCR-induced proliferation of T cells from mice with conditional deletion of STIM1 and STIM2. (B) Failed TCR-induced upregulation of glycolysis (measured by extracellular acidification rate, ECAR) and oxidative phosphorylation (measured by oxygen consumption rate, OCR) in T cells lacking STIM1 and STIM2. (C) Abolished glucose uptake (left), and expression of GLUT1 and hexokinase 2 (middle and right) by STIM1 and STIM2 deficient T cells after TCR stimulation. FK506 (tacrolimus) is an inhibitor of the Ca2+ regulated phosphatase calcineurin. From: Vaeth M et al. Immunity 2017 47:664-679.
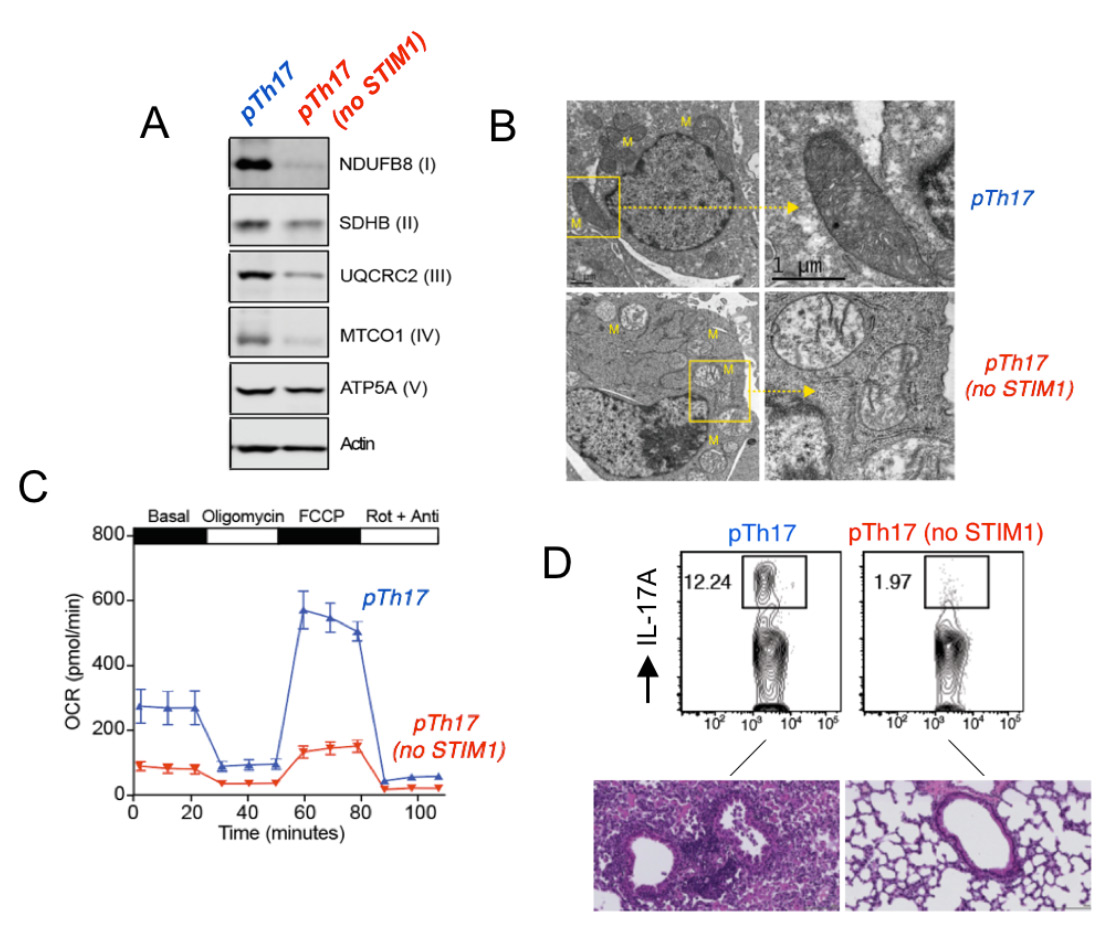
STIM1 and Ca2+ influx are essential for mitochondrial respiration and pathogenic function of Th17 cells. (A) Impaired expression of electron transport chain proteins in pathogenic Th17 cells (pTh17) lacking STIM1. (B) Abnormal mitochondrial ultrastructure with loose cristae in STIM1-deficient pTh17 cells. (C) Near abolished mitcohondrial respiration in STIM1 deficient pTh17 cells measured by Seahorse flux analysis. (D) Strongly reduced IL-17A production by STIM1-deficient pTh17 cells (top) and protection from severe pTh17-induced pulmonary inflammation. From: Kaufmann U. et al. Cell Metabolism 2019 29:1104-1118.
Characterization of novel ion channels and transporters in immune function.
The importance of ion channels for immune function in humans is evident from inherited loss-of-function (LOF) mutations in ion channel genes that cause immunodeficiency (1). LOF mutations in ORAI1 and STIM1 cause CRAC channelopathy, and LOF mutations in the gene encoding the Mg2+ transporter MAGT1 result in XMEN syndrome, severely impairing T and NK cell function. In addition, mutations in the Cl− channel LRRC8A and the Zn2+ transporter ZIP7 were shown to perturb human B cell development. As we learn more about the roles of ICTs in immune cells, this list will surely expand. Compared to other organs, ion channels in the immune system are a neglected topic, especially when compared to excitable cells such as neurons and cardiomyocytes.The reasons are manifold and include the common misunderstanding that immune cells, which are nonexcitable, have no obvious need for ion channels. Nothing could be further from the truth, as ion channels regulate the function of every cell in the body by controlling the cells' membrane potential, Ca2+ signaling, proliferation, apoptosis and many more functions. The mammalian genome encodes several hundred ion channels, transporters, exchangers, pumps and their regulatory proteins that collectively regulate the movement of Ca2+, Mg2+, Zn2+, K+, Na+, Cl−, and other ions across membranes. Of these, about 10-15 have well established roles in immune cell function and immunity based on solid genetic evidence demonstrated by null mutations of human or mouse genes (2,3). Additional evidence comes from pharmacological and gene expression studies. The Feske lab is undertaking a systematic effort to discover new ion channels that regulate immune cell function and immune responses. Our work currently focuses on the identification and characterization of ion channels that control T cell function in the context of infection, autoimmunity (with an emphasis on murine models of MS and colitis) and antitumor immunity. We have successfully established the methodologies to conduct forward genetic screens to identify ion channels regulating T cell mediate immunity in vivo. Other areas of interest are ion channels that regulate innate immune responses by myeloid cells. Drugs targeting ion channels in the heart, vasculature and nervous system are among the most potent and best-selling pharmaceuticals worldwide. By contrast, there are currently no approved drugs modulating channel function in immune-related diseases. This is a missed opportunity for the treatment of immunological disorders including autoimmunity, allergy and antitumor immunity. Ion channels are excellent drug targets as many of them are located at the cell surface, which make them accessible to small molecule inhibitors and biologicals.
- Feske S, Vaeth M. Ion Channelopathies of the Immune System. Current Opinion in Immunology 2018; 52:39-50.
- Feske S. Eye on ion channels. Science Signaling 2019;12 (572).
- Feske S, Wulff H, Skolnik EY. Ion channels in innate and adaptive immunity. Annual Rev Immun 2015; 33:291-353.
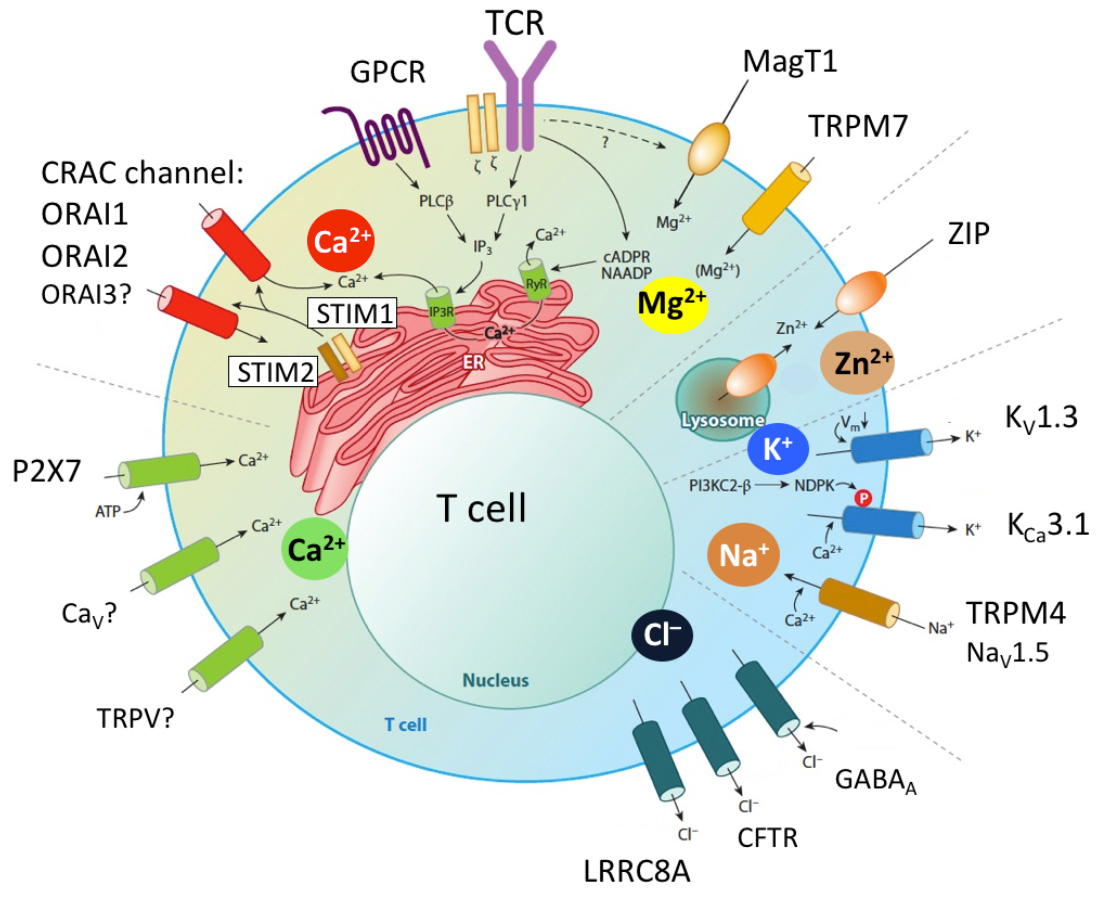
Ion channels in T cells. Antigen sttimulation of the T cell receptor (TCR) or engagement of GPCRs (e.g. chemokine receptors) results in the activation of phospholipase C (PLC) g1 or PLCb, respectively, production of IP3, depletion of Ca2+ from the ER, activation of STIM1 and STIM2 and opening of CRAC channels in the plasma membrane. The CRAC channel is formed by six ORAI proteins of which ORAI1 is the best characterized (whereas the role of ORAI2 and ORAI3 is less well understood). Ca2+ influx mediated by CRAC channels is called store-operated Ca2+ entry (SOCE) because its activation is regulated by the Ca2+ concentration in the ER. Other Ca2+ channels reported have been reported in T cells and P2X receptors (especially P2X7) which are regulated by extracellular ATP. Several Ca2+ permeable TRP channels and voltage gated Ca2+ channels (CaV) have been implicated in T cell function, although their roles in T cells and mechanistic regulation not well defined. Other divalent cation channels in T cells are MagT1 and TRPM7, which mediate Mg2+ influx (although TRPM7 is also permeable to Ca2+) and were shown to regulate T cell function. Several Zn2+ transporters ibelonging to the ZIP and ZnT protein families were reported to regulate intracellular and organellar Zn2+ levels. Their function in T cells is not well understood, despite the important role of Zn2+ in immune function. The influx of Ca2+ (and potentially that of other divalent cations) depends on a negative membrane potential (Vm), which is established by two K+ channels, KV1.3 and KCa3.1, and the Na+ channel TRPM4 that hyper- and depolarize Vm, respectively. All three channels were shown to regulate T cell function in vitro and in vivo. Several Cl– channels have been implicated in T cell function, including the swell activated LRRC8A channel, GABAA receptors, cystic fibrosis transmembrane regulator (CFTR) and Cl–/HCO3– exchanger Ae2.
From: Feske et al. 2015 Ann Rev Imm 33:291-353.

Ion channel and transporter (ICT) expression in immune cell subsets. mRNA expression of > 600 ICTs in murine immune cell subsets. Data from Immgen.org.